|
||||||||
We report the synthesis, crystal structure and electrochemical behaviour of a complex in which the Ph group of the phosphaalkene PhC(H)=PMes* (Mes*: 2,4,6-tri-tert-butylphenyl) is coordinated to a chromium tricarbonyl group. The EPR spectra resulting from electrochemical and chemical reductions are described and the experimental g and hyperfine tensors (31P)T, as determined from the EPR data, are compared with those predicted by DFT calculations for the radical anion (Cr(CO)3, PhC(H)=PMes)·â. The structural changes caused by the addition of an electron to the neutral complex are described, together with an estimation of the contribution of Cr(CO)3 to the stabilization of the radical anion. | ||||||||
|
 |
|||||||
Two new, âuser-friendlyâ derivatives of triptycene containing AsH2 and SiH3 fragments were synthesized. Both solids are crystalline, air-stable compounds characterized by elevated melting points and resistance toward moisture. The highly reactive AsâH and SiâH bonds are protected by the presence of the surrounding phenylene hydrogen atoms, which ensure a remarkable kinetic stabilization of these primary hydrides. After X-ray irradiation of a single crystal of triptycenesilane, a persistent silyl radical was trapped and characterized. | ||||||||
 |
|
|||||||
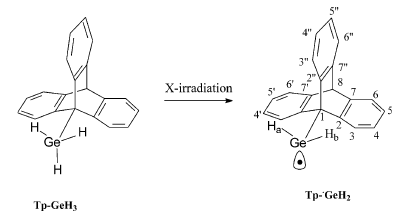
X-irradiation of single crystals of TpâGeH3 (Tp: triptycene) led to the trapping of the radical TpââGeH2. The angular variations of the resulting EPR spectra were recorded at 300 and 77 K. The drastic temperature dependence of the spectra was caused by both a strong anisotropy of the g-tensor and a rotation of the âGeH2 moiety around the CâGe bond. The determination of the EPR tensors as well as the analysis of this motion required to take the presence of disorder in the crystal into account. In accordance with DFT calculations, TpââGeH2 is shown to be pyramidal and to adopt, in its lowest energy structure, a staggered conformation. Rotation around the CâGeH2 bond is blocked at 90 K and is almost free above 110 K. The experimental barrier, obtained after simulation of the EPR spectra as a function of the rotational correlation time, is equal to 1.3 kcal molâ1, which is slightly inferior to the barrier calculated by DFT (3.6 kcal molâ1). Calculations performed on TpâCH3, TpâGeH3 and TpââGeH2 show that the rotation barrier ÎErot around the CâGe bond drastically decreases by passing from the germane precursor to the germanyl radical and that ÎErot increases by passing from the germane to its carbon analogous. Structural parameters involved in these barrier differences are examined. | ||||||||
|
||||||||
The paper shows the possibilities of the complementary use of the density matrix formalism for the simulation of the anisotropic EPR spectra and the DFT potential energy surface calculations to obtain a detailed picture of the motions of radical molecules. The combined approach is illustrated by a comparative EPR study of three phosphorus derivatives of barrelene. Three compounds were chosen as the model molecules for the observation of different temperature dependent dynamics of radical fragment. Each molecule based on the same barrelene skeleton has a different set of substituents which by influencing the local chemical environment are likely to modify the internal dynamics. The temperature dependent EPR spectra are simulated by means of the density matrix formalism and the geometry of radicals are calculated with DFT. The motion is described in terms of rotational barriers, DFT calculated energy profiles and hypothetical intramolecular distortions. These two approaches lead to a similar microscopic picture of the intramolecular radical motion. | ||||||||
|
||||||||
The g, 31P and 1H hyperfine tensors of the dibenzobarrelene phosphinyl radical, trapped in an X-irradiated single crystal of dibenzobarrelene phosphine, were estimated at 45 and 300 K. They indicate that among the three locations of the phosphinyl hydrogen expected from DFT calculations, only two are occupied at 40 K and that the third one remains practically vacant, even at 300 K. The temperature dependence of the EPR spectrum was simulated by assuming jumps between two PâH bond orientations (energy barrier ~= 0.5 kcal molâ1) which correspond to the conformation of the PH2 moiety in the only rotamer present in the dibenzobarrelene phosphine crystal. | ||||||||
|
|
||||||||
A new phosphine, the diphenyldibenzobarrelenephosphine 2, was designed to study the barrier to rotation of the PâH group around the Cââ¢P bond. After homolytic scission of a PâH bond by radiolysis, the EPR spectrum of the resulting phosphinyl radical, trapped in a single crystal of 2, was studied at 77 K and at room temperature. The directions of the 31P hyperfine eigenvectors were compared with the bond orientations of the undamaged compound as determined from its crystal structure. The temperature dependence of the EPR spectrum was analyzed by using the density matrix formalism; this showed that interaction between the phosphinyl hydrogen and the phenyl ring bound to the ethylenic bond is determinant for explaining the potential energy profile. DFT investigations are consistent with these experimental results. | ||||||||





